Фенилкетонурия — наследственное заболевание, связанное с нарушением обмена веществ. Оно может вызвать серьезные последствия для здоровья ребенка при отсутствии своевременной диагностики и лечения. В статье рассмотрим фенилкетонурию, причины, типы наследования, основные симптомы и методы лечения. Эти сведения помогут родителям лучше понять диагноз своего ребенка и принять необходимые меры для его здоровья.
Фенилкетонурия – что это за болезнь?
Фенилкетонурия, известная также как болезнь Феллинга, представляет собой серьезное заболевание, впервые описанное в 1934 году норвежским исследователем Феллингом. В ходе своих исследований он обследовал группу детей с умственной отсталостью и обнаружил у них наличие фенилпирувата в моче – вещества, образующегося при распаде аминокислоты фенилаланина, которая не может быть расщеплена в организме пациентов. Фенилкетонурия является врожденным нарушением обмена веществ и была одной из первых открытых болезней подобного рода.
Фенилкетонурия – это наследственное заболевание, связанное с нарушением обмена аминокислоты фенилаланина. При отсутствии необходимого фермента, отвечающего за его переработку, фенилаланин накапливается в организме, что может привести к серьезным неврологическим нарушениям. Эксперты подчеркивают, что заболевание возникает из-за мутации в гене, отвечающем за синтез фермента фенилаланингидроксилазы.
Лечение фенилкетонурии заключается в строгой диете, которая ограничивает потребление продуктов, богатых фенилаланином, таких как мясо, молочные продукты и орехи. Важно, чтобы родители начали соблюдать диету как можно раньше, чтобы предотвратить развитие осложнений. Также рекомендуется регулярное медицинское наблюдение и контроль уровня фенилаланина в крови. Современные подходы к лечению включают использование специальных заменителей белка, что позволяет детям получать необходимые питательные вещества без риска для здоровья.
https://youtube.com/watch?v=FCPLqburyTo
Фенилкетонурия – тип наследования
Болезнь Феллинга представляет собой наследственное заболевание, связанное с хромосомными и генетическими изменениями, которое передается от родителей к детям. Основным виновником данной патологии является ген, расположенный на 12-й хромосоме. Этот ген отвечает за выработку печеночного фермента фенилаланин-4-гидроксилазы, который необходим для превращения фенилаланина в тирозин — вещество, важное для нормального функционирования организма.
Фенилкетонурия передается как рецессивный признак. Около 2 % населения являются носителями данного дефектного гена, однако сами они не страдают от фенилкетонурии. Заболевание проявляется только в том случае, если оба родителя передают ребенку этот ген, что происходит с вероятностью 25 %. Если фенилкетонурия наследуется как рецессивный признак, и жена является гетерозиготной, а муж — гомозиготным по нормальному аллелю, то вероятность того, что их дети будут здоровыми носителями гена фенилкетонурии, составляет 50 %.
| Аспект | Описание | Важность |
|---|---|---|
| Что такое фенилкетонурия (ФКУ)? | Наследственное метаболическое заболевание, при котором организм не может правильно расщеплять аминокислоту фенилаланин. | Ранняя диагностика и лечение критически важны для предотвращения необратимых повреждений. |
| Причины возникновения ФКУ | Мутация в гене, отвечающем за выработку фермента фенилаланингидроксилазы (ФАГ). Передается по аутосомно-рецессивному типу. | Понимание генетической природы помогает в консультировании семей и планировании беременности. |
| Симптомы и последствия без лечения | На начальных этапах симптомы отсутствуют. Без лечения приводит к накоплению фенилаланина в крови и тканях, вызывая тяжелые неврологические нарушения, задержку умственного развития, судороги, микроцефалию, поведенческие проблемы. | Подчеркивает необходимость неонатального скрининга и строгого соблюдения диеты. |
| Диагностика ФКУ | Неонатальный скрининг (тест пяточки) проводится всем новорожденным на 3-5 день жизни. Измеряется уровень фенилаланина в крови. | Позволяет выявить заболевание до появления симптомов и начать лечение. |
| Лечение ФКУ (диета) | Основной метод лечения – строгая диета с низким содержанием фенилаланина на протяжении всей жизни. Исключаются или ограничиваются продукты, богатые белком (мясо, рыба, молочные продукты, яйца, бобовые, орехи). | Позволяет контролировать уровень фенилаланина и предотвращать развитие осложнений. |
| Специальные продукты и смеси | Использование специализированных безбелковых продуктов и аминокислотных смесей, не содержащих фенилаланин, но обеспечивающих организм всеми необходимыми питательными веществами. | Обеспечивает полноценное питание при соблюдении диеты. |
| Мониторинг и контроль | Регулярный контроль уровня фенилаланина в крови, консультации с диетологом и неврологом. | Позволяет корректировать диету и отслеживать состояние здоровья ребенка. |
| Прогноз при своевременном лечении | При ранней диагностике и строгом соблюдении диеты дети с ФКУ развиваются нормально, без значительных отклонений. | Дает надежду семьям и подчеркивает эффективность лечения. |
Интересные факты
Фенилкетонурия (ФКУ) — это наследственное заболевание, связанное с нарушением обмена веществ, при котором организм не может перерабатывать аминокислоту фенилаланин. Вот несколько интересных фактов об этом заболевании:
-
Генетическая природа: Фенилкетонурия вызывается мутацией в гене, отвечающем за производство фермента фенилаланин-гидроксилазы. Это фермент необходим для превращения фенилаланина в тирозин. Если этот процесс нарушен, уровень фенилаланина в крови повышается, что может привести к серьезным неврологическим нарушениям.
-
Скрининг новорожденных: В большинстве стран проводится обязательный скрининг новорожденных на фенилкетонурию. Это позволяет выявить заболевание на ранней стадии и начать лечение, что значительно снижает риск развития умственной отсталости и других осложнений.
-
Диетическое лечение: Основным методом лечения ФКУ является строгая диета с низким содержанием фенилаланина. Это включает в себя исключение из рациона продуктов, богатых белком, таких как мясо, рыба, яйца и молочные продукты. Вместо этого используются специальные формулы и продукты, обогащенные необходимыми питательными веществами, чтобы обеспечить нормальное развитие ребенка.
Эти факты подчеркивают важность ранней диагностики и соблюдения диеты для управления фенилкетонурией и предотвращения серьезных последствий для здоровья.
https://youtube.com/watch?v=1cLYndK9yPQ
Формы фенилкетонурии
Изучая, у кого может возникнуть фенилкетонурия и что собой представляет это заболевание, чаще всего упоминается классическая форма патологии, которая наблюдается примерно в 98% случаев. Остальные случаи составляют кофакторная фенилкетонурия, вызванная нарушением синтеза или восстановления активной формы тетрагидробиоптерина. Этот компонент является кофактором для нескольких ферментов, и без него их активность не может проявляться.

Фенилкетонурия – причины
Болезнь Феллинга представляет собой патологическое состояние, возникающее в результате мутаций в гене, что приводит к недостатку или полному отсутствию фенилаланин-4-гидроксилазы. Это вызывает накопление фенилаланина и его неполных продуктов расщепления в тканях и физиологических жидкостях организма. Часть избытка фенилаланина преобразуется в фенилкетоны, которые выводятся с мочой, что и дало название данной болезни.
Нарушения обмена веществ оказывают наиболее серьезное влияние на головной мозг. Это приводит к токсическому воздействию на его ткани, нарушает процессы жирового обмена, вызывает сбои в миелинизации нервных волокон и снижает уровень образования нейромедиаторов. В результате запускаются патогенетические механизмы, которые могут привести к задержке умственного развития у ребенка.
https://youtube.com/watch?v=n5Gz4V7AERU
Фенилкетонурия – симптомы
При рождении ребенок с данным заболеванием выглядит совершенно здоровым, и лишь через 2-6 месяцев начинают проявляться первые признаки. Фенилкетонурия начинает давать о себе знать, когда в организме малыша накапливается фенилаланин, поступающий с грудным молоком или искусственными смесями. На начальных этапах могут наблюдаться такие, пока еще не специфические симптомы:
- повышенная вялость или, наоборот, беспокойство;
- беспричинные крики;
- мышечная дистония;
- частые срыгивания;
- судороги;
- нарушения сна.
Кроме того, у таких детей кожа, волосы и глаза имеют более светлый оттенок по сравнению с другими членами семьи, что связано с нарушением синтеза пигмента меланина. Еще один характерный признак, который могут заметить врачи или внимательные родители, – специфический «мышиный» запах, возникающий из-за выделения фенилаланина с мочой и потом.
Более выраженные клинические проявления становятся заметными примерно к полугоду, после введения первого прикорма:
- трудности с фокусировкой взгляда на отдельных предметах;
- безразличие к окружающему;
- отсутствие мимики и улыбки;
- тремор рук и другие.
Также можно заметить физические отклонения: уменьшенный размер головы, выдающаяся вперед верхняя челюсть, задержка в росте. Дети с этим заболеванием поздно начинают держать голову, ползать, садиться и вставать. Характерна особая поза при сидении – поза «портного», когда руки постоянно согнуты в локтях, а ноги – в коленях. Если лечение не начато к трем годам, симптомы становятся более выраженными.

Фенилкетонурия – диагностика
Фенилкетонурия у детей часто диагностируется уже в роддоме, что дает возможность своевременно начать лечение и предотвратить возникновение серьезных необратимых последствий. На 4-5 день после рождения у новорожденных берут капиллярную кровь натощак для выявления ряда тяжелых генетических заболеваний, среди которых находится и фенилкетонурия. Если выписка из роддома произошла раньше, анализ проводится в поликлинике по месту жительства в течение первых 10 дней жизни.
Учитывая, что в редких случаях могут быть получены ложные результаты, диагноз не устанавливается на основании первого анализа. Для подтверждения наличия заболевания назначается ряд дополнительных исследований, включая:
- анализ мочи для выявления фенилпирувата;
- количественное определение фенилаланина и тирозина в плазме крови;
- оценка активности печеночных ферментов;
- электроэнцефалография и магнитно-резонансная томография головного мозга.
Генетический дефект, вызывающий данное заболевание, может быть выявлен у плода с помощью инвазивной пренатальной диагностики. Для этого берутся образцы клеток из ворсинок хориона или амниотической жидкости, после чего проводится ДНК-анализ. Рекомендуется проводить такое исследование в семьях с повышенным риском возникновения заболевания, особенно если в семье уже есть ребенок с фенилкетонурией.
Фенилкетонурия – лечение
При диагностировании фенилкетонурии у новорожденных необходимо, чтобы за состоянием пациентов следили специалисты, такие как генетик, педиатр, невропатолог и диетолог. Тем, кто знаком с фенилкетонурией, будет ясно, что основным методом лечения является соблюдение диеты с ограничением фенилаланина. Кроме того, могут быть назначены медикаменты, массаж, лечебная физкультура, а также психолого-педагогические подходы, направленные на социализацию ребенка и подготовку его к обучению.

Фенилкетонурия – диета
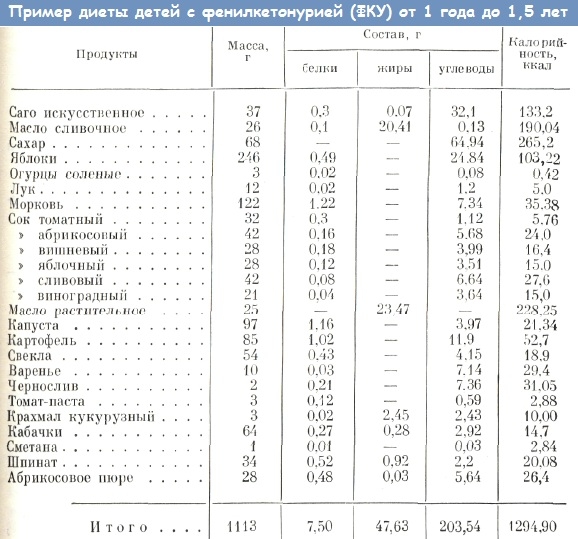
При установлении диагноза «фенилкетонурия» ребенку сразу же назначается специальная диета. Из питания исключаются продукты, содержащие высокое количество белка, такие как мясо, рыба, молочные изделия, бобовые и орехи. Потребность в белках компенсируется с помощью специализированных диетических смесей и других продуктов, содержащих берлофен – полусинтетический гидролизат белка, который полностью свободен от фенилаланина (например, Тетрафен, Лофеналак, Нофелан). Пациенты могут употреблять безбелковый хлеб, макароны, крупы, муссы и подобные продукты. Грудное вскармливание осуществляется в ограниченных количествах.
Строгое соблюдение диеты и регулярный мониторинг уровня фенилаланина в крови в течение первых 14-15 лет жизни помогают предотвратить развитие умственных нарушений. В дальнейшем рацион может быть немного расширен, однако многие эксперты советуют придерживаться специального питания на протяжении всей жизни. Кофакторная форма фенилкетонурии не поддается лечению с помощью диеты, а корректируется только с помощью препаратов тетрагидробиоптерина.
Фенилкетонурия – препараты для лечения
Лечение фенилкетонурии у детей включает в себя использование различных медикаментов, среди которых:
- ноотропные препараты (Пирацетам, Церебролизин);
- витамины группы В;
- минеральные комплексы;
- средства, способствующие улучшению тканевого обмена (АТФ, Рибоксин);
- препараты, улучшающие микроциркуляцию (Трентал, Пентоксифиллин).
Фенилкетонурия – прогноз в отношении жизни и болезни
Родители, знакомые с генетическим заболеванием фенилкетонурия, сегодня имеют шанс вырастить здорового ребенка, если будут строго придерживаться рекомендаций врачей. В отсутствие необходимого лечения прогноз для пациентов с фенилкетонурией оказывается крайне неблагоприятным: такие люди, как правило, доживают до 30 лет, страдая от серьезных умственных нарушений и различных функциональных проблем.
Фенилкетонурия – профилактика и генетическое консультирование
Фенилкетонурия (ФКУ) – это наследственное заболевание, связанное с нарушением обмена аминокислоты фенилаланина. Это состояние возникает из-за недостатка фермента фенилаланингидроксилазы, который отвечает за превращение фенилаланина в тирозин. При отсутствии этого фермента фенилаланин накапливается в организме, что может привести к серьезным неврологическим нарушениям и умственной отсталости. Профилактика ФКУ и генетическое консультирование играют ключевую роль в предотвращении развития заболевания у новорожденных и их последующем здоровье.
Профилактика фенилкетонурии начинается с ранней диагностики. В большинстве стран новорожденные проходят скрининг на ФКУ в первые дни жизни. Этот тест позволяет выявить заболевание до появления первых симптомов, что значительно увеличивает шансы на успешное лечение и предотвращение осложнений. Если у ребенка выявляется повышенный уровень фенилаланина, его направляют на дальнейшее обследование и консультацию с генетиками.
Генетическое консультирование является важным этапом для семей, в которых уже есть случаи фенилкетонурии. Специалисты помогают оценить риск передачи заболевания будущим детям, объясняют механизмы наследования и предлагают варианты для планирования беременности. Если оба родителя являются носителями гена, отвечающего за ФКУ, вероятность рождения ребенка с этим заболеванием составляет 25%. В таких случаях могут быть предложены методы предимплантационной диагностики, которые позволяют отобрать эмбрионы без мутации.
Кроме того, генетическое консультирование может включать в себя информацию о возможностях лечения и диетических рекомендациях для детей с ФКУ. Своевременное вмешательство и соблюдение строгой диеты, низкой по содержанию фенилаланина, могут значительно улучшить качество жизни и предотвратить развитие серьезных осложнений. Важно, чтобы родители понимали, что ранняя диагностика и соблюдение рекомендаций врачей являются ключевыми факторами в управлении заболеванием.
Таким образом, профилактика фенилкетонурии и генетическое консультирование являются важными аспектами в борьбе с этим заболеванием. Они помогают не только выявить заболевание на ранних стадиях, но и обеспечить поддержку семьям, столкнувшимся с этой проблемой, что в конечном итоге способствует улучшению здоровья и качества жизни детей с ФКУ.
Вопрос-ответ
Почему появляется фенилкетонурия?
Причина заболевания связана с нарушением обмена незаменимой аминокислоты фенилаланина, приводящим к повышению ее уровня в крови, тканях и биологических жидкостях. Избыток фенилаланина токсичен для нервной системы и при длительном воздействии вызывает в ней необратимые дегенеративные изменения.
Какая причина фенилкетонурии?
Фенилкетонурия (ФКУ) вызывается мутацией в гене, отвечающем за синтез фермента фенилаланин-гидроксилазы, который необходим для превращения аминокислоты фенилаланина в тирозин. При недостатке этого фермента фенилаланин накапливается в организме, что может привести к серьезным неврологическим нарушениям и умственной отсталости, если не проводить своевременное лечение.
Можно ли вылечить фенилкетонурия?
Болезнь фенилкетонурия особенная тем, что ее можно вылечить и практически полностью устранить симптомы, в том случае, если она диагностирована до или сразу после рождения.
Советы
СОВЕТ №1
Обязательно проконсультируйтесь с врачом-генетиком, если в вашей семье есть случаи фенилкетонурии. Раннее выявление заболевания у новорожденного может значительно улучшить качество жизни и предотвратить серьезные осложнения.
СОВЕТ №2
Следите за диетой вашего ребенка. При фенилкетонурии необходимо строго ограничить потребление белка, так как аминокислота фенилаланин может накапливаться в организме и вызывать токсические эффекты. Используйте специальные смеси и продукты, рекомендованные врачом.
СОВЕТ №3
Регулярно проходите обследования и тесты на уровень фенилаланина в крови. Это поможет контролировать состояние вашего ребенка и своевременно корректировать лечение и диету.
СОВЕТ №4
Обучите себя и близких основам ухода за ребенком с фенилкетонурией. Знание о заболевании, его симптомах и методах лечения поможет вам лучше поддерживать своего малыша и обеспечивать ему полноценную жизнь.